
By Mike Thompson, CEO
DyNAbind is a DNA-Encoded Library company, but a huge part of our unique technology platform is built on exploiting principles of fragment-based drug discovery (FBDD). In this blog post, I will give some context for what FBDD is, why it is an attractive strategy for discovery, and how DyNAbind’s approach offers the best of both worlds with a hybrid DEL and FBDD platform. I’ll try to be concise but leave plenty of links to more substantial sources for anyone that wants to go down the rabbit hole.
While biologics and other big molecules (e.g. macrocycles, peptides) are of continually growing interest, the majority of drug discovery approaches are still based on classical small molecule approaches. Exact definitions for ‘small molecule’ vary, but a safe place to start is something below 900 Da. In reality, many screening collections try to stay within Lipinski or ‘Rule-of-5’ space (MW < 500, logP < 5, etc.), as these molecules are more likely to be orally bioavailable. FBDD goes even further, building collections around ‘fragment’ molecules which can be said to obey a ‘Rule-of-3’, (MW < 300, logP < 3, etc.).

 Figure 1: The active fragments are first identified using random fragment library screening, and after obtaining the three-dimensional (3D) structure of the fragments binding to the target protein, SBDD (Structured Based Drug Discovery) methods are used to optimize and link these active small molecules in order to produce potential lead compounds.
Figure 1: The active fragments are first identified using random fragment library screening, and after obtaining the three-dimensional (3D) structure of the fragments binding to the target protein, SBDD (Structured Based Drug Discovery) methods are used to optimize and link these active small molecules in order to produce potential lead compounds.
This may not seem like a wild departure from the typical small molecule approach, but there are
actually significant consequences to using these smaller starting points. For one, you get a
significantly more effective sampling of chemical space. There are more theoretical druglike
small molecules than there are stars in the universe. Even DEL, which rose to prominence for
its massive throughput, can barely scratch the surface here.
However, for every heavy (non-hydrogen) atom we remove from a theoretical molecule, the size of chemical space decreases 8-fold. So, via some back-of-the-envelope math, a collection of 150 Da fragments will sample the available chemical space something like 150-200 times better than an equally sized collection of 500 Da small molecules.
There are also perks for the people working downstream in hit-to-lead optimization. Fragment-based hits tend to have better ligand efficiency scores than their small molecule counterparts. In case that’s a new term to you, ligand efficiency is the Gibbs free energy of binding divided by the number of non-hydrogen atoms in the molecule. Hit-to-lead optimization is a delicate balancing act where molecules are tuned to maximize affinity and specificity while also keeping a clean physchem and ADME profiles. Give your chemists a highly ligand-efficient starting point and they will love you for it.
To me, though, the most exciting thing about FBDD is what it can do for you in enabling new target space. This will probably be its own blog post at some point, but the fast explanation is that the drug target landscape is dramatically changing. Less than 20% of the human genome consists of what are called ‘druggable’ targets, or proteins with deep, generally hydrophobic binding pockets with enough differentiation from other proteins that a specific binder can be found. The remaining 80+% of the genome consists of ‘undruggable’ targets which have been resistant to classical discovery methods. A common issue with these proteins is the lack of those traditional deep binding pockets, featuring rather broad, flat and relatively featureless surface interactions. Given that traditional small molecule approaches are designed to work in compact, hydrophobic cavities, it shouldn’t be surprising that they underperform in these conditions, with less than 0.01% of studied protein-protein interactions (PPIs) able to be modulated by small molecules.
Fragments, on the other hand, have been showing great promise here, with numerous fragment-based PPI binders reported. Multiple FBDD drugs for PPIs are already on the market, with at least another 10 in clinical trials. The fragments generally work by grabbing onto ‘hot spots’ in the surface interactions, single residue regions which have a strong contribution to potential binding. The increased 2-dimensionality and bare bones, just-the-core, construction of fragments gives them a better chance for grabbing these hot spots and creating the foundation for a downstream drug molecule against these traditionally ‘undruggable’ proteins.
Sounds good, right? Fragments are absolutely great tools, but you have to remember that there is no magic bullet in drug discovery. Every tool in the box has its advantages, disadvantages and preferred use cases, and FBDD is no different. So what are the downsides here? For one, many of the classic fragment discovery methods are cumbersome, e.g. NMR or X-ray crystallography. Also, fragments tend to have very weak initial interaction strength, even down to the moderate or high millimolar range. That means you’re looking at high sample consumption, and in some cases you have to start worrying about solubility issues.
The weak interactions also mean that work needs to be done to increase the potency downstream. This is typically done by growing, linking or merging the fragments. There are plenty of successful examples of this being done, but growth usually requires structural knowledge, while merging or linking requires the identification of fragments which bind sufficiently proximal sites for feasible connection.
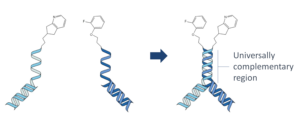 By combining DEL with FBDD concepts, we’ve grabbed the best of both approaches. DyNAbind’s patented Dynamic Library system consists of two fragment sub libraries which contain a universally
By combining DEL with FBDD concepts, we’ve grabbed the best of both approaches. DyNAbind’s patented Dynamic Library system consists of two fragment sub libraries which contain a universally
complementary DNA annealing region. When we mix the two sublibraries together, we get an exponential growth of compound diversity as we form structures that present two fragments to the target simultaneously. The annealing region of DNA is intrinsically
unstable, so that the fragment pairs are constantly reshuffling until their configurations are stabilized by binding to the target. So, as we incubate the library, the library self-optimizes to recycle weakly or non-binding structures into more potent binders.
This unique DEL approach gives us the flexibility and power of FBDD while minimizing the drawbacks. The screening assay used is the same target agnostic method that DEL always uses, so applying it to any target is straightforward. Thanks to the DNA tags, solubility is no longer a concern for the fragments. And finally, by looking at dual-fragment structures, we are already delivering appropriate pairs of fragments for linking together. Our proprietary on-DNA hit validation technology means that we can get binding affinity data for the fragment pairs without doing any resynthesis or linking chemistry, so we can rapidly triage the hits, confirm superadditivity effects and make smart decisions about where to put development resources.
We’ve had great success applying this technology to a number of classically undruggable targets like PPIs and transcription factors. If you’re struggling with a protein that’s resistant to old-school discovery methods, then get in touch! Our Dynamic Library platform might be just the thing to kick-start your development.
References:
A general-purpose introduction to fragment-based discovery: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6930586/
Dan Erlanson’s practical fragments blog: http://practicalfragments.blogspot.com/
A review on some of the different targets hit by fragment-based discovery: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00180/full
A DyNAbind case study on our fragment-enabled discovery and follow-up linking: https://pubs.rsc.org/en/content/articlelanding/2019/cc/c9cc01429b