
By Mike Thompson, CEO
Over the last few decades, approaches to drug discovery and development have become markedly more rational. It really wasn’t that long ago that we were screening random natural product extracts hoping for an active substance. The advent of modern molecular biology along with data-driven insight have led to direct screening methodologies against recombinant protein using small molecule collections optimized for oral bioavailability. Even more recently, the successful sequencing of the human genome in the early 2000s peeled back the curtain to offer a more complete view than ever before of the biological basis of diseases, and what the actual target landscape looks like. In this post I’ll comment on some of the more striking things we’ve learned from this, what the emerging target space looks like, and what it means for drug discovery strategy moving forward.
 To start, despite the relatively large number of FDA approved drugs, there is a surprisingly limited number of protein classes for which small molecule drugs have been developed. In fact, 70% of drugs only target four protein classes: GPCRs, ion channels, kinases, and nuclear receptors. With only a few exceptions, these broad classes of proteins are classically druggable targets, i.e., they contain deep binding pockets with a well-defined shape where the natural substrate binds. In most cases, new drugs were designed based on the structure of the endogenous ligands, thus mainly acting as competitive inhibitors.
To start, despite the relatively large number of FDA approved drugs, there is a surprisingly limited number of protein classes for which small molecule drugs have been developed. In fact, 70% of drugs only target four protein classes: GPCRs, ion channels, kinases, and nuclear receptors. With only a few exceptions, these broad classes of proteins are classically druggable targets, i.e., they contain deep binding pockets with a well-defined shape where the natural substrate binds. In most cases, new drugs were designed based on the structure of the endogenous ligands, thus mainly acting as competitive inhibitors.
To the best of our knowledge, the complete collection of FDA approved drugs targets about 850 different proteins, while Hopkins and Groom estimated the ‘druggable genome’ (i.e., proteins which are believed to be capable of modulation via traditional small molecules approaches) to contain about 3000 proteins, or about 10% of the total human genome. So, what do we take away from this? First, while we are not in immediate danger of running out of druggable targets, they are a finite resource. Second, it’s important to remember that ‘capable of modulation with small molecules’ does not translate to ‘will be drugged.’ Less than 5% of drug development projects result in approved drugs, with many failing due to target-related issues, e.g., lack of binder specificity due to binding site homology or lack of efficacy against the actual disease, potentially due to unknown redundant systems.
Fig. 1. Types of binding surfaces
So, while the established approach has been productive, our increased understanding of protein modulation as well as rapid advances in genomics, proteomics and structural biology have made it clear that an increased focus is needed on ‘undruggable’ targets along with correspondingly innovative discovery approaches. In many cases, the binding sites of these proteins consist of large and flat surfaces instead of deep pockets, a feature that renders inviable the design of potent inhibitors by mimicking the natural compounds. One notable example that has revolutionized cancer therapy are the recent advances in treatment of tumours containing the KRAS G12C mutation by the 2021 approval of Sotorasib, a covalent binder that allosterically disrupts the GTP-mediated activation of this historically undruggable protein. The KRAS inhibitor market is expected to be worth over 4.5 billion USD by 2028.
Perhaps no better target class exists for expanding the potential druggable space than protein-protein interactions (PPIs). These interacting systems influence a huge number of physiological and pathological processes, with the size of the so-called connectome estimated at over 650,000 cases. However, as with KRAS, these proteins lack the hydrophobic ligand pockets that favor traditional discovery methodology, rather utilizing broad, relatively featureless surfaces for interaction. As I discussed in our previous blog post, fragment-based drug discovery (FBDD) has emerged as a utile method for hitting PPIs. Many success cases are already in the literature, such as Bcl-2 with venetoclax, one of the first PPI drugs, and more recently by the development of other promising anticancer agents like bromodomain inhibitors or modulators of XIAP/caspase-9.
More recently, the concept of protein degradation by heterobifunctional small-molecule proteolysis-targeting chimeras (PROTACs) has also gained traction as a strategy for hitting undruggable proteins. PROTACs consist of a bifunctional molecule that engages on one
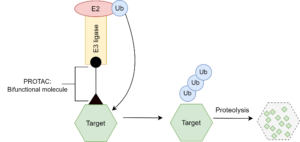 side, an E3 ubiquitin ligase, and the protein to be degraded on the other. Thanks to this approach, several inhibitors have been found to show good biological activity against a broad variety of disease-associated proteins, from estrogen and androgen receptors (ER and AR) to cereblon (CRBN) and Von Hippel–Lindau disease tumor suppressor protein (VHL), among others. PROTACs are attractive as a binder needs only high affinity to the target, with no functionality required. On the other hand, they face the same problems as PPIs, in that finding a strong enough binder for the surfaces can be a challenge.
side, an E3 ubiquitin ligase, and the protein to be degraded on the other. Thanks to this approach, several inhibitors have been found to show good biological activity against a broad variety of disease-associated proteins, from estrogen and androgen receptors (ER and AR) to cereblon (CRBN) and Von Hippel–Lindau disease tumor suppressor protein (VHL), among others. PROTACs are attractive as a binder needs only high affinity to the target, with no functionality required. On the other hand, they face the same problems as PPIs, in that finding a strong enough binder for the surfaces can be a challenge.
Fig 2: PROTAC Mechanism
That’s where DyNAbind jumps in.
 Our proprietary Dynamic Library technology was designed from the ground up to hit challenging target surfaces. By offering dynamically recombining fragment pairs to the target simultaneously, we take advantage of the better surface-interacting potential of fragments, while offering synergistic binding that can massively increase initial affinity compared to classical FBDD. We’ve successfully deployed our libraries on numerous ‘undruggable’ PPI and PROTAC systems and are always ready to bring in another. If you’re looking over the edge of druggable target space, get in touch with us!
Our proprietary Dynamic Library technology was designed from the ground up to hit challenging target surfaces. By offering dynamically recombining fragment pairs to the target simultaneously, we take advantage of the better surface-interacting potential of fragments, while offering synergistic binding that can massively increase initial affinity compared to classical FBDD. We’ve successfully deployed our libraries on numerous ‘undruggable’ PPI and PROTAC systems and are always ready to bring in another. If you’re looking over the edge of druggable target space, get in touch with us!
DyNAbind is ready to be your partner in opening new target opportunities.
References:
- The Evolving Druggability and Developability Space: Chemically Modified New Modalities and Emerging Small Molecules (https://pubmed.ncbi.nlm.nih.gov/31900602/).
- New Frontiers in Druggability (https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b00586)
- The druggable genome: Twenty years later (https://www.frontiersin.org/articles/ 10.3389/fbinf.2022.958378/full)
Taking Aim at the Undruggable (https://ascopubs.org/doi/full/10.1200/EDBK_325885)